Traducción de ‘Fibrous Dysplasia/McCune-Albright Syndrome-GeneReviews’ Texto original
Puedes descargar la guía médica completa en castellano en formato PDF aquí, también tienes un resumen aquí.
Índice
- Autores
- Características clínicas
- Diagnóstico
- Enfermedades genéticas relacionadas
- Manejo terapéutico y seguimiento
- Consejo genético
- Referencias
Autores
Dra. Alison M Boyce, MD
Pediatra endocrino especializada en la evaluación de enfermedades esqueléticas en niños y adolescentes con especial énfasis en la Displasia Fibrosa/Síndrome de McCune-Albright. Realiza su trabajo investigador en los Institutos Nacionales de la Salud (NIH) en Bethesda, Maryland, EEUU. Forma parte del equipo de la sección de Enfermedades Esqueléticas y de la Homeostasis Mineral (del inglés Skeletal Disorders and Mineral Homeastasis Section, SDMHS).
Dr. Pablo Florenzano, MD
Endocrinólogo especializado en la evaluación y tratamiento de enfermedades óseas en adultos. Realiza su trabajo investigador en la Pontificia Universidad Católica de Chile.
Dr. Luis Fernández de Castro, PhD
Biólogo del SDMHS del NIH. Realiza investigación básica y traslacional sobre enfermedades óseas y de metabolismo mineral.
Dr. Michael T Collins, MD
Endocrinólogo especializado en enfermedades raras del hueso y del metabolismo mineral. Jefe del SDMHS del NIH.
Traducción al castellano:
Dra. Diana Ovejero Crespo
Endocrina especializada en la evaluación de enfermedades esqueléticas. Actualmente realiza su investigación en el Instituto de Investigaciones Médicas del Hospital del Mar (IMIM) en Barcelona.
Características clínicas
La DF/MAS es secundaria a la activación mosaica de mutaciones en el gen GNAS. Este gen codifica la proteína Gsα, la cual es una subunidad de los receptores acoplados a proteínas G que está involucrada en la vía de señalización del AMP cíclico (cAMP). La mutación puede estar presente en tejidos que deriven tanto del ectodermo, del mesodermo como del endodermo, y frecuentemente afecta a la piel, el esqueleto, y algunos órganos endocrinos. No obstante, dado que la señalización por Gsα está presente en prácticamente todos los tejidos del cuerpo, otras localizaciones pueden verse afectadas también.
La presentación clínica es extremadamente variable: desde hallazgos incidentales sin clínica asociada, a letalidad neonatal. La variabilidad clínica entre individuos es muy elevada, tanto en el número de tejidos afectos como en el grado en el que dichos tejidos están afectados. La manifestación de la enfermedad depende del momento embrionario en el que la mutación tiene lugar, los tejidos afectos, y del papel de Gsα en estos tejidos. Generalmente, las mutaciones que se producen en un momento temprano del desarrollo resultan en una mayor extensión de la enfermedad.
Piel
Manchas café con leche. Estas manchas son frecuentes y suelen ser la primera manifestación de la enfermedad (al nacimiento o al poco de nacer). NO existe una correlación entre el tamaño de las manchas y la extensión de la enfermedad ósea/endocrina. Tampoco existe una relación entre la distribución de las manchas y la localización de la DF.
Displasia Fibrosa
La DF muestra una distribución mosaica en el esqueleto y puede afectar a cualquier parte y cualquier combinación del esqueleto craneofacial, axial y/o apendicular. Los huesos que se afectan más frecuentemente son la base del cráneo y los fémures proximales (Kelly et al 2008).
La DF tiene un amplio espectro de manifestación: desde lesiones aisladas asintomáticas halladas de manera incidental a una enfermedad poliostótica severa que afecte a prácticamente todos los huesos del esqueleto resultando en una incapacitación severa con pérdida de visión, audición y movilidad.
Las lesiones de DF suelen manifestarse durante los primeros años de vida y tienden a aumentar de tamaño durante la infancia. La vasta mayoría de las lesiones se pueden detectar antes de los 10 años de edad, siendo muy raro que aparezcan nuevas lesiones después de los 15 años (Hart et al 2007). En la edad adulta, las lesiones de DF suelen ser menos activas (Kuznetsov et al 2008).
La afectación clínica y la evolución de la DF dependen de la localización y extensión de la enfermedad:
Esqueleto apendicular:
- En niños, la presentación típica de DF en el esqueleto apendicular con DF incluye el dolor, la cojera, y/o las fracturas patológicas.
- El desarrollo de fracturas recurrentes y deformidades progresivas puede resultar en dificultades en la marcha y pérdida de movilidad.
Región cráneofacial:
- La DF en esta región puede manifestarse como un “bulto” o como asimetría facial.
- La expansión de las lesiones cráneofaciales puede derivar en una deformidad facial progresiva (ver Figura 2B), y en raras ocasiones (normalmente asociada a secreción excesiva de hormona de crecimiento) a la pérdida de visión y/o audición secundarias a la compresión de los nervios ópticos y canales auditivos externos, respectivamente (Cutler et al 2006, Boyce et al 2018).
Columna vertebral:
- La afectación de las vértebras es frecuente y puede derivar en escoliosis que en raras ocasiones puede ser grave, progresiva e incluso letal (Leet et al 2004b).
- La falta de tratamiento de la escoliosis progresiva es una de las pocas causas de morbimortalidad temprana en la DF.
El dolor óseo es una complicación muy común de la DF. Aunque el dolor pueda manifestarse a cualquier edad, lo más típico es que no esté presente durante la infancia, aparezca durante la adolescencia, y progrese durante la vida adulta (Kelly et al 2008).
Los quistes óseos aneurismáticos son lesiones rápidamente expansivas llenas de fluido que se originan en lesiones de DF preexistente. El método diagnóstico de detección para estos quistes es la resonancia magnética (RMN). Los síntomas principales incluyen la aparición aguda de dolor intenso y de una deformidad rápidamente expansiva. Si el quiste se localiza en la región cráneofacial y comprime el nervio óptico, podría manifestarse también como pérdida de visión. Así pues, los quistes óseos anuerismáticos tienen asociada una alta morbilidad (ver Manejo terapéutico).
La transformación maligna de la DF es una complicación rara. En varias ocasiones se ha reportado una asociación entre la transformación maligna de la DF con el antecedente de radioterapia (Ruggieri et al 1994). El exceso de hormona de crecimiento también podría constituir un factor de riesgo (Salenave et al 2014).
Apariencia radiográfica clásica de la displasia fibrosa
Las radiografías en el esqueleto apendicular suelen mostrar lesiones expansivas con una apariencia en “vidrio esmerilado”, adelgazamiento de la corteza, e irregularidades endosteales (Ver Figura 2A). En la región cráneo-facial, la DF suele aparecer en las radiografías convencionales como lesiones expansivas escleróticas, y en la tomografía computerizada suelen mostrar una apariencia de “vidrio esmerilado” (Ver Figura 2C). Con el envejecimiento, las lesiones de DF en el esqueleto apendicular tienden a adoptar en las radiografías una apariencia esclerótica, mientras que en la región cráneo-facial adoptan una apariencia “quística” (Figura 2D).
Endocrinopatías
Pubertad precoz.
Es muy frecuente en niñas con DF/MAS (~85%). Es habitual que la pubertad precoz sea el síntoma inicial de la enfermedad. Los quistes ováricos recurrentes (Ver Figura 4A) conlleva la producción intermitente de estrógenos lo cual resulta en el desarrollo de senos, aceleración del crecimiento lineal y sangrado vaginal. Durante los intervalos entre la involución de quistes antiguos y la formación de quistes nuevos, el tejido mamario suele involucionar y los niveles de estrógeno sérico retornan a niveles prepuberales. El desarrollo de quistes ováricos suele continuar en la edad adulta, por lo que es frecuente que las pacientes afectas presenten ciclos menstruales irregulares (Lala et al 2007). La torsión ovárica se ha descrito en raras ocasiones en niñas y mujeres con quistes persistentes de gran tamaño (Clark et al 2000).
La pubertad precoz es menos frecuente en niños con DF/MAS (~10-15%), y es secundaria a la producción autónoma de testosterona (Boyce et al 2012a) resultando en un desarrollo puberal progresivo, incluyendo una aceleración en el crecimiento lineal, crecimiento de vello púbico y axilar, la aparición de acné, y el desarrollo de una comportamiento inapropiadamente agresivo y sexual.
Tanto en niños como en niñas con pubertad precoz, la producción autónoma prolongada de esteroides sexuales generalmente culmina en la activación del eje hipotálamo-hipofisario y en el desarrollo de pubertad precoz central.
Fertilidad.
El efecto de la producción de esteroides sexuales sobre la función gonadal y la fertilidad no han sido adecuadamente caracterizados. Las mujeres con DF/MAS pueden tener quistes recurrentes resultando en periodos irregulares durante la edad adulta (Lala et al 2007). Mientras que muchas mujeres de la cohorte del NIH han conseguido embarazos con éxito, es posible que la interrupción de los ciclos ovulatorios pueda disminuir la fertilidad y aumentar en el tiempo hasta la concepción (observación personal de los autores)
Alteraciones testiculares.
Las alteraciones testiculares están presentes en la mayoría de varones con MAS (~85%), y típicamente se manifiestan como macroorquidismo unilateral o bilateral (Boyce et al 2012a). La exploración ecográfica suele mostrar discretas lesiones hiper e hipoecoicas así como microlitiasis, las cuales corresponden a áreas de hiperplasia de células de Leydig y/o Sertoli (Ver Figura 4B).
Se desconoce el potencial de malignización de estas lesiones testiculares, pero los estudios clínicos disponibles indican que es muy bajo (Boyce et al 2012a).
Enfermedad tiroidea.
La afectación de la glándula tiroides es muy frecuente en los pacientes con DF/MAS. Se detectan alteraciones ecográficas tiroideas en aproximadamente la mitad de los individuos con DF/MAS. Estas alteraciones incluyen la presencia de lesiones mixtas solido-quísticas y lesiones sólidas entremezcladas con áreas de tejido de apariencia normal. (Figura 4C y 4D) (Celi et al 2008, Tessaris et al 2012a).
El hipertiroidismo suele afectar al 10-30% de los individuos con DF/MAS y es el resultado de tanto el incremento de producción hormonal como del incremento de la conversión de tiroxina (T4) a triyodotironina (T3) (Celi et al 2008). El hipertiroidismo suele ser leve o moderado, pero en ocasiones puede ser más grave pudiendo incluso llegar a provocar una tormenta tiroidea durante la inducción anestésica prequirúrgica (Lawless et al 1992).
El hipertiroidismo no tratado puede resultar en el adelanto de la edad ósea, en la elevación del remodelado óseo y en fracturas.
La transformación maligna de los tejidos tiroideos afectos puede ocurrir de manera excepcional (Collins et al 2003).
Pérdida urinaria de fosfato mediada por FGF23.
La producción aumentada de la hormona factor de crecimiento fibroblástico 23 (FGF23) en áreas de DF puede resultar en una tubulopatía renal con pérdidas urinarias de fosfato (Collins et al 2001). No obstante, no es frecuente que se detecte hipofosfatemia franca en pacientes con DF dado que la mayor parte del FGF23 producido, es degradado en el tejido displásico (Bhattacharyya et al 2012). El grado de sobreproducción de FGF23 en pacientes con DF suele correlacionarse con el grado de extensión de la enfermedad esquelética, por lo que los casos de hipofosfatemia franca sólo afecta a individuos con una afectación esquelética extensa (Rimunucci et al 2003).
En pacientes con DF/MAS las pérdidas urinarias de fosfato pueden aparecer de manera intermitente durante el transcurso de la vida del paciente, siendo más pronunciadas en periodos de crecimiento esquelético rápido. También es habitual que las alteraciones del fosfato desparezcan a medida que el individuo envejece (Kuznetsov et al 2008).
Los individuos que presenten hipofosfatemia franca tiene un riesgo incrementado de padecer raquitismo, osteomalacia, fracturas y dolor óseo (Lee et al 2004a).
Exceso de producción de hormona de crecimiento.
Aproximadamente un 15-20% de los individuos con DF/MAS son portadores de la mutación GNAS patogénica en la hipófisis anterior lo cual puede resultar en la producción autónoma de hormona de crecimiento (GH). Se estima que el 80% de los individuos que presente exceso de GH también presentarán hipeprolactinemia de manera concomitante (Salenave et al 2014).
El exceso de GH en individuos con DF/MAS suele manifestarse inicialmente con aceleración del crecimiento lineal y pueden llegar a desarrollar características de acromegalia. Clínicamente, el exceso de GH debe diferenciarse de la pubertad precoz y del hipertiroidismo, los cuales también se asocian a una aceleración en el crecimiento lineal.
El exceso de GH no tratado se asocia con la expansión de las lesiones de DF en la región cráneo-facial resultando en macrocefalia y un riesgo aumento en la pérdida de agudeza visual (Boyce et al 2013) (ver Figura 2B).
Hipercortisolismo.
En raras ocasiones, los bebés con DF/MAS pueden debutar con un síndrome de Cushing secundario a un exceso de producción de cortisol por la glándula adrenal fetal (Brown et al 2010, Carney et al 2011). Los síntomas suelen desarrollarse durante el periodo neonatal y pueden llegar a ser muy graves resultando incluso en la muerte. La regresión espontánea de este síndrome de Cushing se ha reportado en aproximadamente la mitad de los supervivientes, presuntamente secundaria a la involución de la glándula fetal adrenal.
Afectación hepática.
- La hepatitis y la colestasis neonatales pueden llegar a ser pronunciadas en bebés, aunque su intensidad suele mermar con la edad, evolucionando a formas leves persistentes (Silva et al 2000, Ikawa et al 2016).
- Se han identificado y reportado adenomas hepáticos portadores de la mutación GNAS patogénica (Gaujoux et al 2014).
- No se han descrito casos de insuficiencia hepática en adultos con DF/MAS.
Afectación grastrointestinal.
- El reflujo gastroesofágico se manifiesta en la infancia y puede ser severo.
- Los pólipos en el tracto gastrointestinal superior han surgido recientemente como un hallazgo común en individuos con DF/MAS (Wood et al 2017).
Afectación pancreática.
Aproximadamente un 15% de los individuos con DF/MAS presentan alteraciones pancreáticas incluyendo:
- Pancreatitis
- Neoplasias mucinosas papilares intraductales (IPMN), las cuales pueden presentar diversos grados de displasia (Gaujoux et al 2014, Wood et al 2017).
- Se ha reportado un caso de un paciente con un carcinoma pancreático derivado de un subtipo de IPMN intestinal (Parvanescu et al 2014).
Mixomas
Se han identificado mixomas intramusculares benignos y asintomáticos de manera incidental.
Alteraciones hematológicas
Se han descrito casos de individuos con DF/MAS con insuficiencia medular con pancitopenia y hematopoyesis extramedular que han requerido una esplenectomía (Mahdi et al 2017, Robinson et al 2018).
Cáncer de mama
Es posible que el riesgo de cáncer de mama en mujeres con DF/MAS esté incrementado y que ocurra en edades más tempranas que en la población general. No obstante, la mutación patogénica en GNAS sólo se ha identificado en aproximadamente la mitad de los tumores de mama que han sido estudiados en mujeres con DF/MAS (Majoor et al 2018a).
Calidad de vida
Varios estudios han demostrado que los individuos con DF/MAS tienen alteraciones en el funcionamiento físico que suele tener una buena correlación con la extensión y gravedad de la enfermedad. No obstante, los individuos afectos suelen preservar el funcionamiento social y emocional. Estos hallazgos tienen relevancia a nivel pronóstico y proporciona alivio a los padres (Kelly et al 2005, Majoor et al 2018b).
Correlación genotipo – fenotipo
No se conocen correlaciones entre un determinado genotipo y el fenotipo.
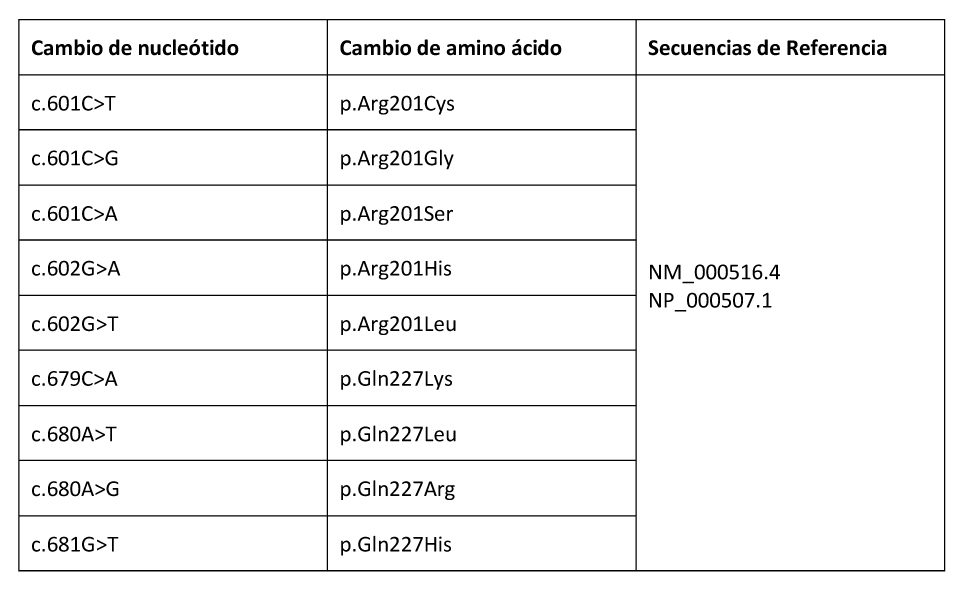
A día de hoy, sólo se han identificado mutaciones en los codones p.Arg201 y p.Gln227 de GNAS. La gravedad de la enfermedad se correlaciona con el grado de mosaicismo y los tejidos afectos.
Nomenclatura
La asociación de mixomas intramusculares con DF/MAS se denomina síndrome de Mazabraud (Cox et al 2017).
Prevalencia
El MAS es una entidad muy rara. Actualmente, no existen datos de prevalencia fiables pero se estima que estará entre los 1:100.000 a 1:1.000.000 nacidos.
Por otro lado, la DF (especialmente la DF monostótica), no es rara. De hecho se estima que constituye hasta un 7% de todos los tumores óseos benignos.
La DF/MAS afecta a ambos sexos y no se ha visto que afecte con mayor frecuencia a ninguna población en particular.
Diagnóstico
Diagnóstico de la displasia fibrosa (DF) y del síndrome de McCune-Albright (MAS) se suele realizar mediante pruebas clínicas, aunque a día de hoy no se han publicado criterios diagnósticos oficiales.
Hallazgos Sugestivos
Se debe sospechar la presencia de DF/MAS en individuos con cualquiera de los siguientes hallazgos:
Piel
Los pacientes pueden presentar unas lesiones cutáneas típicas denominadas manchas “café con leche”.
Los bordes suelen ser irregulares y serrados. En Estados Unidos se conoce este patrón como “Costa de Maine”. La distribución de las lesiones suele respetar la línea media del cuerpo (aunque no siempre) y sigue las denominadas líneas de Blaschko, las cuales reflejan patrones de migración de células embrionarias (ver figura 1).
Figura 1. Lesiones cutáneas
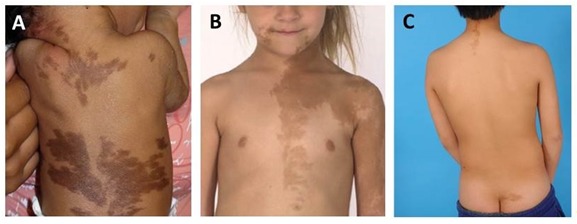
A. Lesiones cutáneas en un recién nacido respetando la línea media del cuerpo y mostrando una distribución que refleja los patrones de la migración de las células embionarias (líneas de Blaschko).
B. Manchas “café con leche” típicas con un aspecto serrado e irregular que afectan al tórax, la cara y el brazo.
C. Manchas “café con leche” de aspecto característico en la nuca y en la hendidura glútea.
Esqueleto
La Displasia Fibrosa (DF) es un trastorno en el que el hueso y la médula ósea normales son sustituidos por un tejido fibroso. Esto conlleva un incremento del riesgo de fracturas, deformidades, deterioro funcional, y dolor.
- La DF puede clasificarse como monostótica (sólo está afectado un hueso), o poliostótica (afectación de más de un hueso).
- La DF puede afectar cualquier parte del esqueleto cráneo-facial, axial o apendicular (ver Figura 2).
Figura 2. Displasia Fibrosa
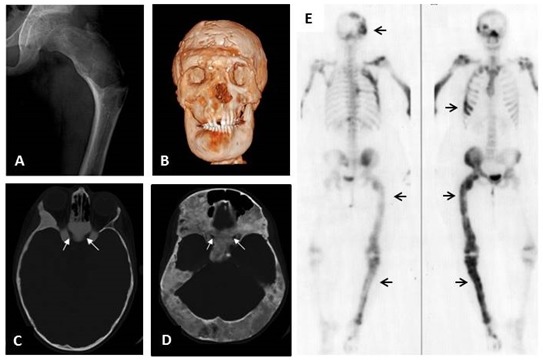
A. Fémur proximal con la típica apariencia radiológica de la DF de “vidrio esmerilado”. El fémur presenta la deformidad en coxa vara denominada en inglés “cayado de pastor” la cual constituye la deformidad esquelética más frecuente en pacientes con DF. B. Imagen de TAC tridimensional reconstruida de un paciente de 26 años con afectación cráneo-facial y secreción excesiva de hormona de crecimiento resultando en macrocefalia y deformidad facial severa. C. Imagen de TAC de una niña de 10 años mostrando la típica apariencia de “vidrio esmerilado” en pacientes jóvenes. Es muy frecuente que los canales ópticos estén recubiertos de DF en aquellos pacientes con DF cráneo-facial (flechas blancas). A pesar de ello, la capacidad visual no se suele afectar. D. Imagen de una mujer de 40 años mostrando hallazgos típicos de la DF cráneo-facial en un individuo de mayor edad en la que se visualiza una apariencia más esclerótica con componentes mixtos quístico-sólidos (flechas blancas). En este caso también hay un recubrimiento de DF alrededor de los nervios ópticos sin afectación visual. E. Gammagrafía ósea con Tecnecio-99 postero-anterior (izquierda) y antero-posterior (derecha), mostrando una captación parcheada en las localizaciones afectadas de DF incluyendo el cráneo, las costillas, el fémur y la tibia (flechas), coherente con la naturaleza mosaica de la enfermedad.
Ante la sospecha de DF, es aconsejable realizar una gammagrafía ósea durante la evaluación radiológica inicial.
Las áreas afectadas que hayan sido identificadas mediante gammagrafía, deberían ser evaluadas más detalladamente mediante radiografías y/o una tomografía computerizada (TAC) craneal dependiendo de la localización y extensión de la enfermedad.
En la Figura 3 se exponen las pruebas de evaluación recomendadas para el diagnóstico de DF.
Figura 3. Pruebas y seguimiento recomendado tras el diagnóstico inicial en individuos con Displasia Fibrosa / Síndrome de McCune-Albright

Endocrino
Se pueden identificar las siguientes alteraciones:
- Pubertad precoz de origen periférico (independiente de la secreción de gonadotropinas).
- Lesiones testiculares incluyendo la hiperplasia por células de Leydig y/o Sertoli. Pueden o no estar asociadas a pubertad precoz de origen periférico. Estas lesiones tienen una apariencia ecográfica característica (ver Figura 4B).
- Lesiones tiroideas con apariencia ecográfica característica, asociadas o no a hipertiroidismo autoinmune (ver Figuras 4C y 4D).
- Secreción excesiva de hormona de crecimiento.
- Pérdida renal de fosfato secundaria a la secreción excesiva de factor de crecimiento fibroblástico 23 (FGF23) con o sin hipofosfatemia asociada.
- Hipercortisolismo neonatal.
Figura 4. Imágenes ecográficas en el Síndrome de McCune-Albright
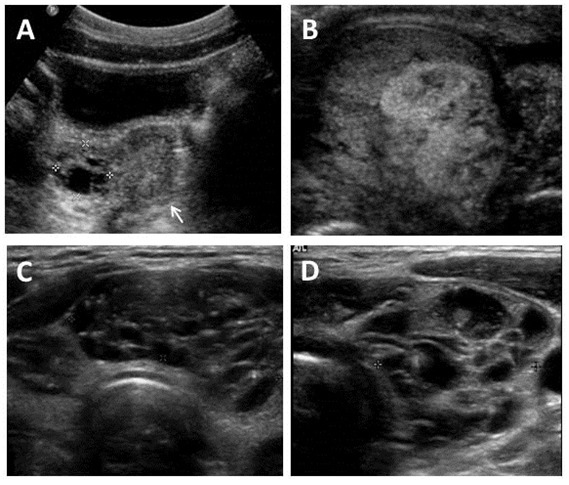
A. Ecografía pélvica en una niña de 7 años mostrando un quiste ovárico complejo unilateral (delimitado por las cruces). El útero tiene un tamaño prepúber (flecha).
B. Ecografía testicular en un adulto mostrando una lesión heterogénea con elementos mixtos sólidos y quísticos.
C&D. Hallazgos tiroideos ecográficos típicos del Síndrome de McCune-Albright incluyendo la apariencia heterogénea y quística (de “queso suizo”).
Establecimiento del diagnóstico
El diagnóstico de DF/MAS puede establecerse en individuos que presenten 2 o más hallazgos clínicos típicos. Para confirmar el diagnóstico en aquellos individuos en los que el único hallazgo clínico sea la DF monostótica, se requerirá la identificación de las variantes patogénicas en el gen GNAS (codones p.Arg201 (>95% de los casos, siendo especialmente frecuente la sustitución por cistina e histidina (p.Arg201Cys y p.Arg201His)) y p.Gln227 (<5% de los casos siendo especialmente frecuente la sustitución por Leucina (p.Gln22Leu)).
El análisis mediante métodos basados en la secuenciación por PCR tiene una mayor sensibilidad si se realiza sobre tejido afecto:
- 80% en tejido afecto.
- 20-30% en linfocitos de sangre periférica.
Notas:
- La detección de variantes patogénicas, es decir de mutaciones, depende del nivel de mosaicismo en el tejido y de la sensibilidad de la técnica. La tasa de identificación de la mutación de GNAS p.Arg201 usando técnicas de PCR estándar es más elevada cuando los tejidos analizados son órganos y menor cuando se realiza sobre especímenes cutáneos (Lumbros et al 2004). La capacidad para detectar mosaicismo afecta la tasa de detección de la técnica (ver Sección de Genética Molecular y Tabla 8).
- El análisis directo de los codones puede realizarse secuenciando los exones 8 y 9 de GNAS. Si se identificasen variantes en el gen GNAS diferentes de aquellas ya reportadas y asociadas a FD/MAS, se interpretarían como variantes de significado incierto.
- La proteína Gsα se expresa en casi todos los tejidos procedentes de los alelos GNAS materno y paterno. No obstante, GNAS es un locus complejo donde el “imprinting” del gen puede resultar en transcritos y fenotipos alternativos.
- Cuando se emplean primers modificados (ácido nucleico peptídico)(Bianco et al 2000) y a su vez se emplea la técnica de Next Generation Sequencing (NGS), la tasa de detección de la variante p.Arg201 es del prácticamente 100% en tejidos afectos y hasta en un 75% en los leucocitos de sangre periférica (Narumi et al 2013).
Enfermedades genéticas relacionadas
Mientras que la DF/MAS son secundarias a mutaciones activadoras en GNAS, la pérdida de función en GNAS pueden dar lugar a una serie de fenotipos característicos. Asimismo, dado que GNAS es un gen improntado (del inglés “genetic imprinting”), el fenotipo varía según el origen materno o paterno del alelo mutado y del grado de impronta genética que tiene lugar en un tejido determinado. En la Tabla 1 se muestran una lista de fenotipos secundarios a variantes inactivadoras patogénicas en GNAS (ver trastornos de la activación GNAS).
Se han identificado mutaciones típicas de la DF/MAS del gen GNAS en varios tumores esporádicos (incluyendo tumores de hipófisis, páncreas, mama, y colon) no asociados a ningún otro rasgo de DF/MAS. La predisposición a desarrollar este tipo de tumores no es hereditaria. Para más información ver la sección “Genética Molecular”.
Tabla 1. Trastornos genéticos secundarios a variantes germinales que causen la inactivación de GNAS
| Fenotipo | Variante GNAS | Referencia OMIM |
|---|---|---|
| Pseudohipoparatiroidismo | Inactivación heterocigota del alelo paterno de GNAS | 612463 |
| Pseudohipoparatiroidismo Ia | Inactivación heterocigota del alelo materno de GNAS en los exones 1-12. | 603233 |
| Pseudohipoparatiroidismo Ib | Defecto de impronta genética secundaria a la deleción heterocigota de los elementos de regulación del locus GNAS materno1. | 603233 |
| Pseudohipoparatiroidismo Ic | Inactivación heterocigota del alelo materno de GNAS en el exón 13. | 612462 |
| Heteroplasia osificante progresiva | Inactivación heterocigota del alelo paterno de GNAS | 166350 |
(1) El pseudohipoparatiroidismo Ib puede ser también causado por la deleción heterocigota del gen STX16.
Diagnóstico diferencial
Neurofibromatosis tipo 1 (NF1)
La NF1 y la DF/MAS comparten varias características clínicas incluyendo la presencia de manchas café con leche y alteraciones esqueléticas. A diferencia de las manchas café con leche típicas del MAS que suelen tener bordes irregulares, las manchas en la NF1 suelen tener bordes lisos. Respecto a alteraciones esqueléticas, en la NF1 es habitual el desarrollo de la cifoescoliosis, displasia esfenoidal, adelgazamiento cortical y arqueamiento de los huesos largos, especialmente de la tibia, que puede evolucionar a pseudoartrosis. Otros rasgos distintivos de la NF1 son los tumores del sistema nervioso central como los neurofibromas y los gliomas ópticos, los hamartomas pigmentados del iris, y las pecas axilares. La NF1 es causada por mutaciones heterocigotas en el gen NF1 y es de herencia autosómica dominante.
Síndrome hipofosfatémico cutáneo esquelético (CSHS)
Este síndrome se caracteriza por la presencia de nevus epidérmicos y/o melanocíticos, una displasia esquelética mosaica, e hipofosfatemia mediada por FGF23. Otros órganos también pueden verse afectados como los ojos, el cerebro, y el sistema vascular entre otros (Ovejero et al 2016). Similarmente a la DF/MAS, es una enfermedad genética mosaica, pero en este caso las mutaciones patogénicas ocurren en los genes HRAS y NRAS.
Lesiones esqueléticas fibro-óseas
Hay lesiones que pueden tener una apariencia radiológica y/o histológica similar a la DF. Estas lesiones suelen ser únicas, no están asociadas con otras alteraciones extra-esqueléticas, y no son portadoras de la mutación GNAS patogénica.
Tumor de células gigantes: Son lesiones adquiridas con rasgos histopatológicos parecidos a la DF, incluyendo la proliferación de las células estromales de la médula ósea y la presencia de células gigantes multinucleadas. Los tumores de células gigantes son habitualmente benignos pero pueden producir destrucción ósea a nivel local y muy raramente, metastatizar. Fibromas osificantes: Son lesiones benignas que habitualmente afectan a la mandíbula y maxilar como una masa expansiva, firme e indolora. Los fibromas osificantes suelen ser más agresivos que la DF cráneo-facial y se tratan mediante la extirpación quirúrgica.
Displasia osteofibrosa
Estas lesiones aparecen habitualmente en niños menores de 10 años y afectan con mayor frecuencia a la tibia anterior. Los niños afectos suelen manifestar una tumefacción localizada indolora, y en raras ocasiones, con fracturas o deformidad. Las radiografías muestran lesiones radiolúcidas bien definidas con un borde esclerótico característico alrededor de la superficie intracortical.
Querubismo
Se caracteriza por el crecimiento progresivo de lesiones fibro-óseas principalmente en la mandíbula y el maxilar. Suele manifestarse en la infancia con un agrandamiento bilateral y simétrico de la parte inferior de la cara proporcionando un aspecto típico de “querubín” en la que los ojos parecen estar mirando hacia arriba. La deformidad facial progresa durante la infancia y la pubertad, tras la cual ocasionalmente involuciona espontáneamente. En la mayoría de los casos, el querubismo es consecuencia de mutaciones heterocigotas en el gen SH3BP2 y sigue un patrón de herencia autosómico dominante.
Manejo terapéutico y seguimiento
Evaluaciones tras el diagnóstico inicial
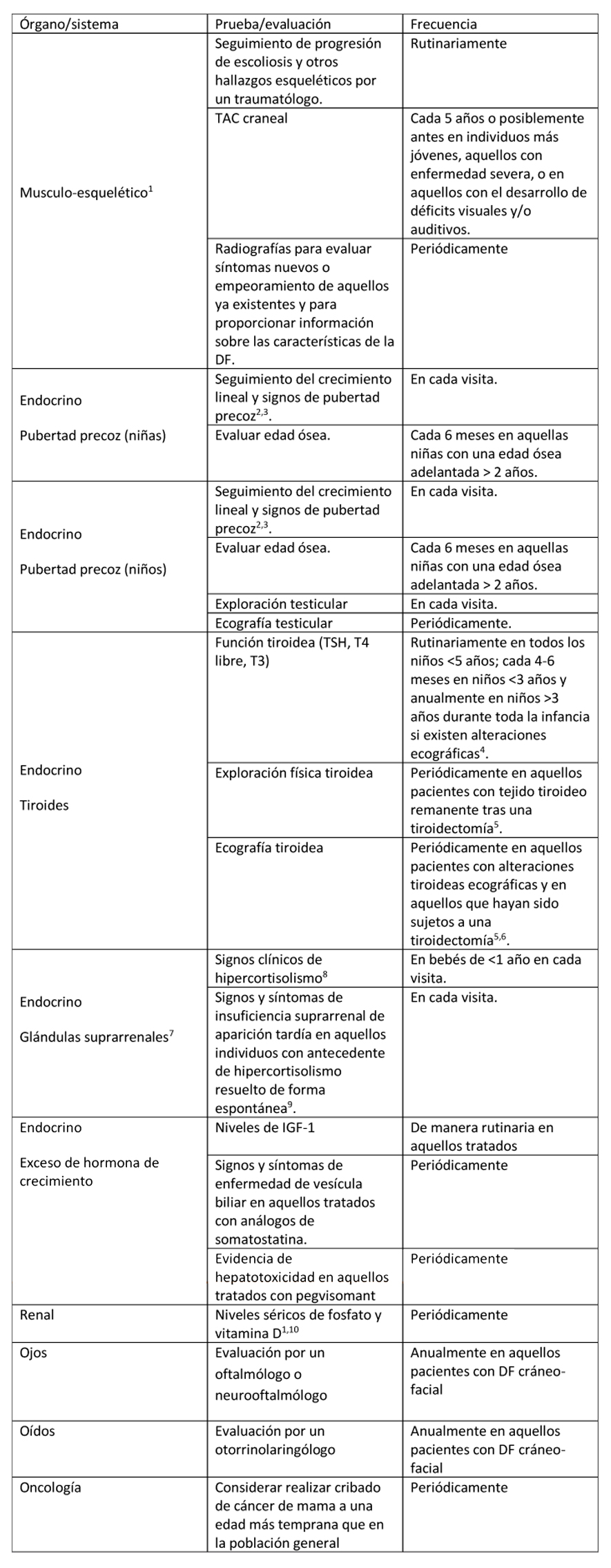
Tras confirmarse el diagnóstico de DF/MAS, se debe realizar un estudio para determinar la extensión de la enfermedad en los individuos afectos. Los autores de esta revisión recomiendan realizar los estudios detallados en la Tabla 2 si no se han realizado hasta ese momento.
Tabla 2. Evaluaciones recomendadas tras el diagnóstico inicial en individuos con Displasia Fibrosa/Síndrome de McCune-Albright
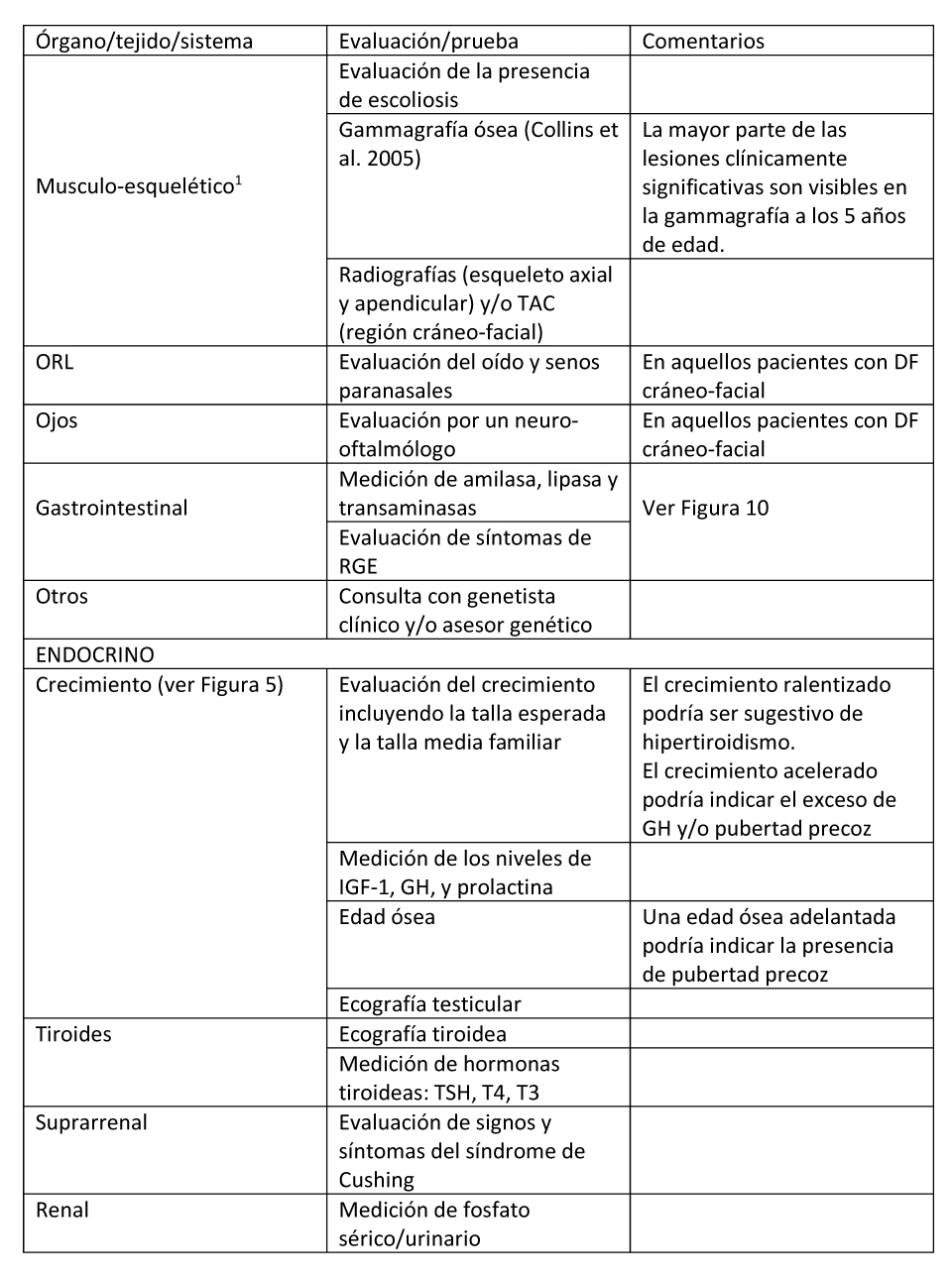
(1) Ver Figura 3 en la que se exponen los estudios recomendados para evaluar el esqueleto tras el diagnóstico. ORL: sistema otorrinolaringológico; RGE: reflujo gastro-esofágico.
Figura 5. Pruebas y seguimiento recomendado para la evaluación del exceso de hormona de crecimiento en individuos con Displasia Fibrosa / Síndrome de McCune-Albright

Figura 6. Pruebas y seguimiento recomendado para la evaluación de trastornos gonadales en niñas con Displasia Fibrosa/Síndrome de McCune-Albright

Figura 7. Pruebas y seguimiento recomendado para la evaluación gonadal de niños con Displasia Fibrosa/Síndrome de McCune-Albright

Figura 8. Pruebas y seguimiento recomendado para la evaluación de alteraciones tiroideas en pacientes con Displasia Fibrosa/Síndrome de McCune-Albright
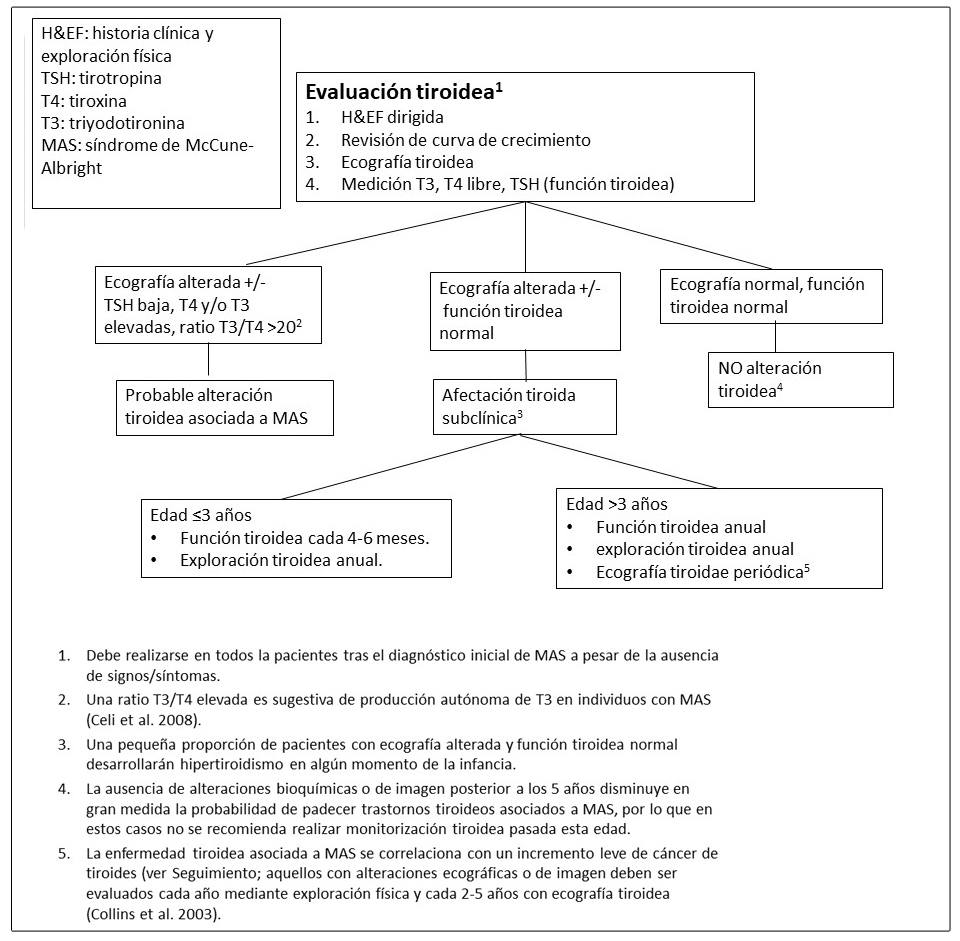
Figura 9. Evaluaciones recomendadas para la disfunción de las glándulas suprarenales en individuos con Displasia Fibrosa/Síndrome de McCune-Albright
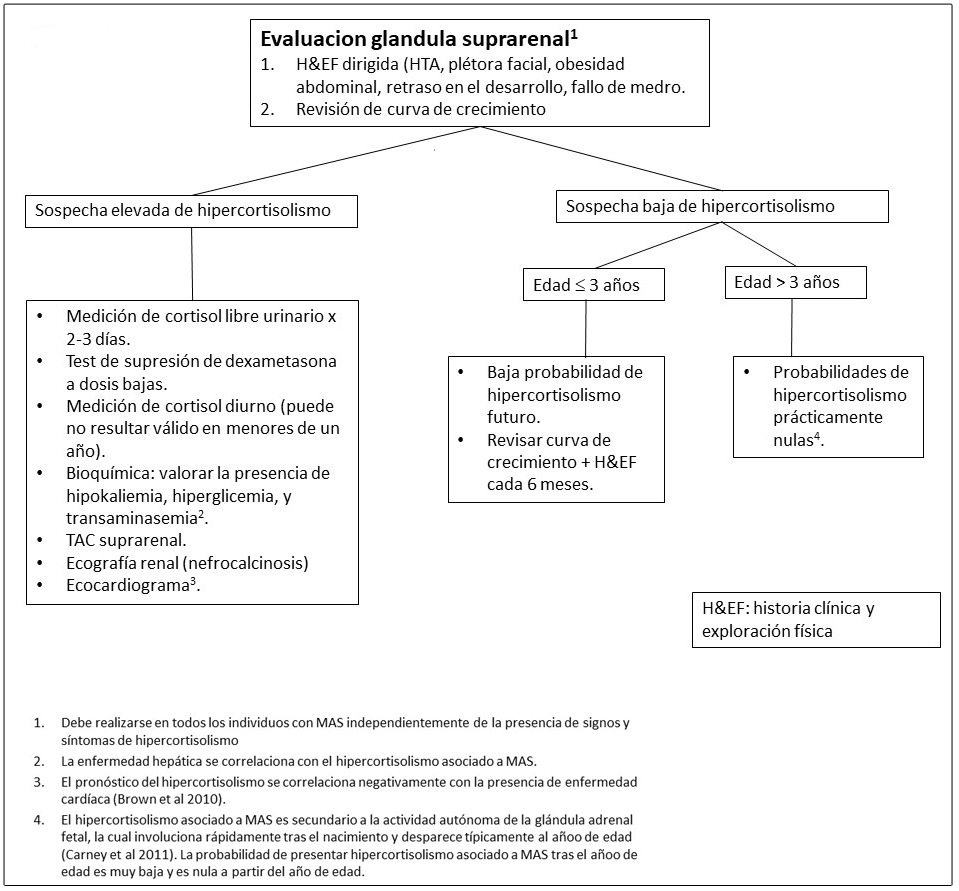
Figura 10. Evaluaciones recomendadas para trastornos gastrointestinales en individuos con Displasia Fibrosa/Síndrome de McCune-Albright
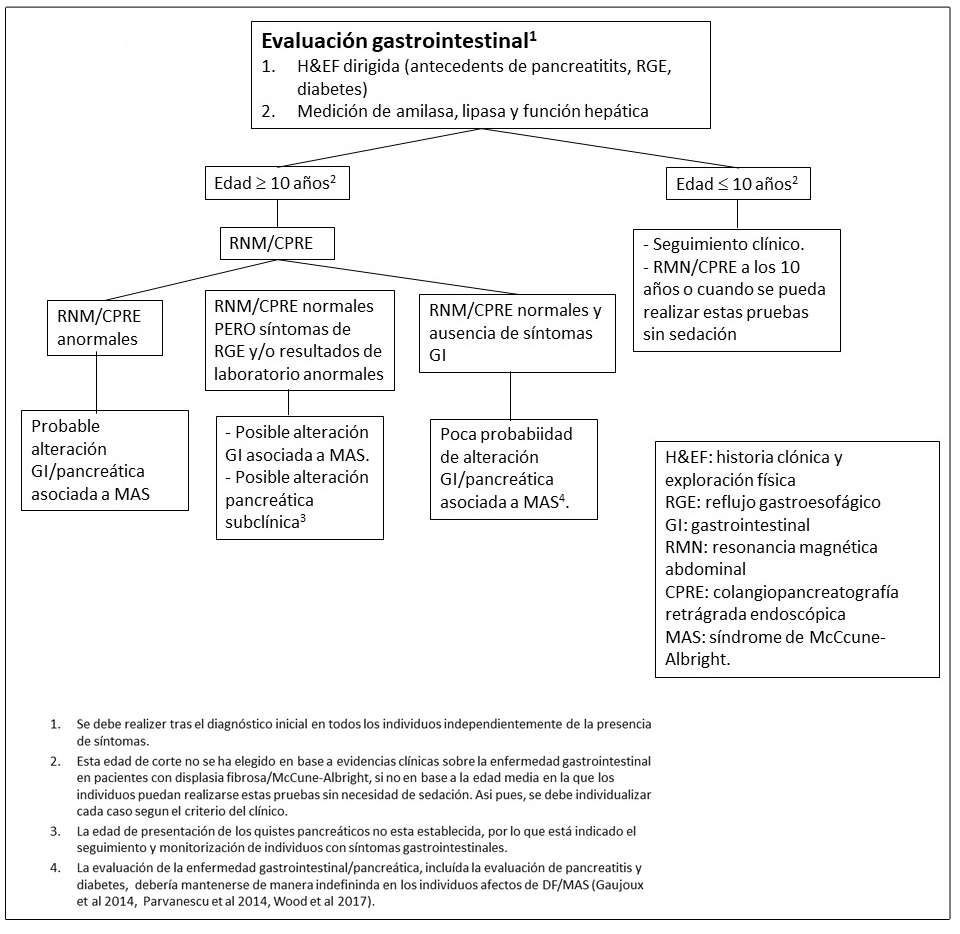
Tratamiento de las manifestaciones
El manejo más efectivo de la enfermedad se suele conseguir mediante la colaboración de un equipo médico multidisciplinar incluyendo endocrinólogos, traumatólogos, oftalmólogos, otorrinolaringólogos, cirujano endocrino, genetistas, médicos rehabilitadores y fisioterapeutas.
Tabla 3. Evaluaciones Recomendadas tras el Diagnóstico Inicial en Individuos con Displasia Fibrosa/Síndrome de McCune-Albright
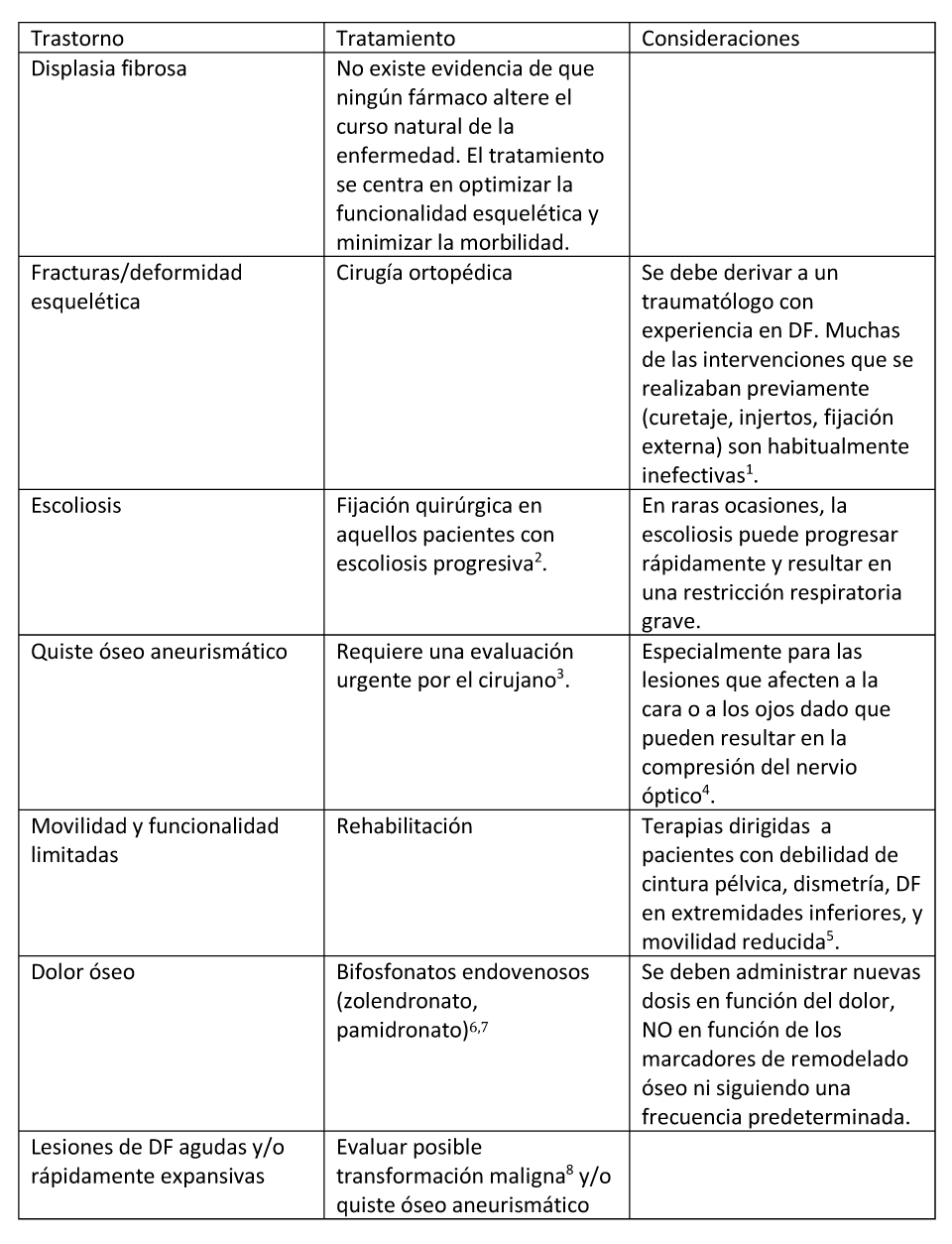
- Stanton et al 2012, Leet et al 2016
- Leet et al 2004b, Mancini et al 2009
- Leet et al 2012, Manjila et al 2013
- La descompresión profiláctica del nervio óptico para reducir el riesgo de pérdida visual está contraindicada ya que de hecho aumenta el riesgo de pérdida visual: Leet et al 2002, Cutler et al 2006, Amit et al 2011.
- Paul et al 2014.
- El tratamiento con alendronato oral no es efectivo para el control del dolor óseo (Boyce et al 201)
- hh
- hh
Figura 11. Manejo clínico recomendado para individuos con Displasia Fibrosa

Tabla 4. Tratamiento de Endocrinopatías en individuos con Displasia Fibrosa/Síndrome de McCune-Albight
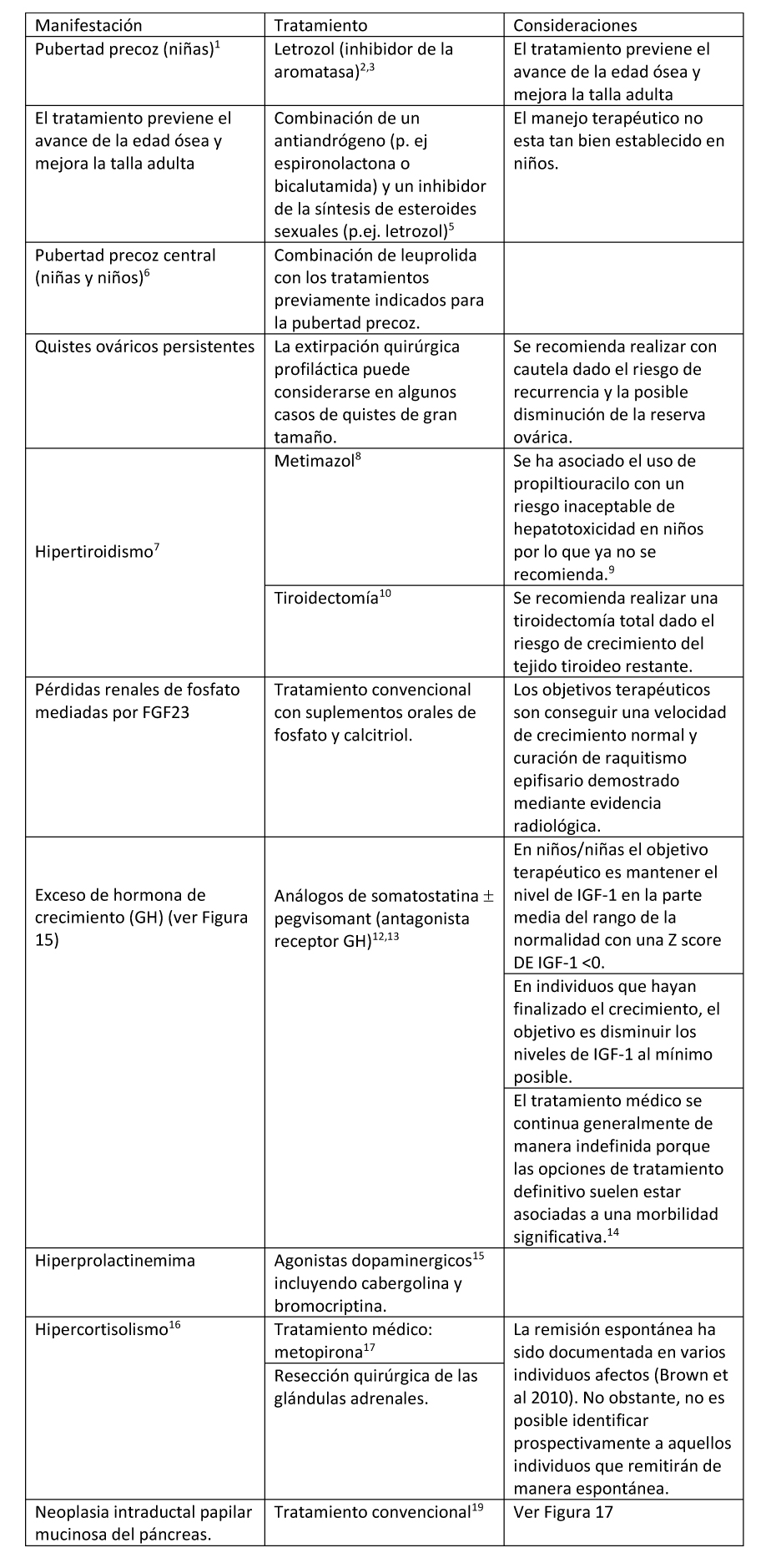
- Ver Figura 12. La mayoría de las niñas tendrán una disminución en el número de sangrados menstruales durante el tratamiento.
- Feuillan et al (2007)
- El tratamiento con letrozol produjo efectos beneficiosos prolongados sobre la maduración esquelética, velocidad de crecimiento, y talla final adulta (Estrada et al 2016).
- Ver Figura 13. La pubertad precoz no es frecuente en varones.
- Boyce et al 2012a.
- Dada la exposición prematura a esteroides sexuales (ver Descripción Clínica), la pubertad precoz central puede presentarse a pesar de que el/la niño/a tuvieran un buen control de la pubertad precoz periférica.
- Ver Figura 14. La radioablación debe evitarse (Ver Agentes/Situaciones a evitar).
- Tessaris et al 2012a.
- Ross et al 2016.
- Se debe realizar con un cirujano endocrino con mucha experiencia para minimizar el riesgo de complicaciones y optimizar los resultados.
- Los marcadores de remodelado óseo (p.ej. la fosfatasa alcalina), puede estar elevados de manera constitutiva y no son un marcador útil de respuesta al tratamiento para la enfermedad esquelética.
- Boyce et al 2013, Salenave et al 2014.
- El tratamiento con radiación puede ser efectivo en casos refractarios, pero se ha asociado a la transformación maligna de la DF cráneo-facial (Hansen &Moffat 2003, Liu et al 2011).
- La cirugía puede ser técnicamente compleja o imposibilitada por la presencia de DF cráneo-facial. Asimismo, dada la infiltración difusa de las células somatotrofas (células pituitarias productoras de GH), los individuos afectos requieren una hipofisectomía total si se intervienen mediante cirugía. Esto resulta en un hipopituitarismo total (Vortmeyer et al 2012).
- Esta clase de medicamentos puede tener un efecto en el tratamiento de exceso de GH en aquellos individuos con elevaciones leves de GH e IGF-1, con o sin hiperprolactinemia asociada (Katznelson et al 2014).
- Ver Figura 16; es difícil establecer guías de práctica clínica especificas dada la rareza del síndrome de Cushing neonatal.
- Se prefiere sobre el ketoconazol en niños con alteraciones hepáticas.
- La decisión de realizar o retrasar la adrenalectomía se debe individualizar, teniendo en cuenta la severidad de la enfermedad, la capacidad de controlar los niveles de cortisol con el tratamiento farmacológico, y los efectos potenciales del hipercortisolismo continuo sobre el desarrollo neuronal.
- Tanaka et al 2012.
Figura 12. Manejo clínico recomendado para la pubertad precoz en niñas con Displasia Fibrosa/Síndrome de McCune-Albright
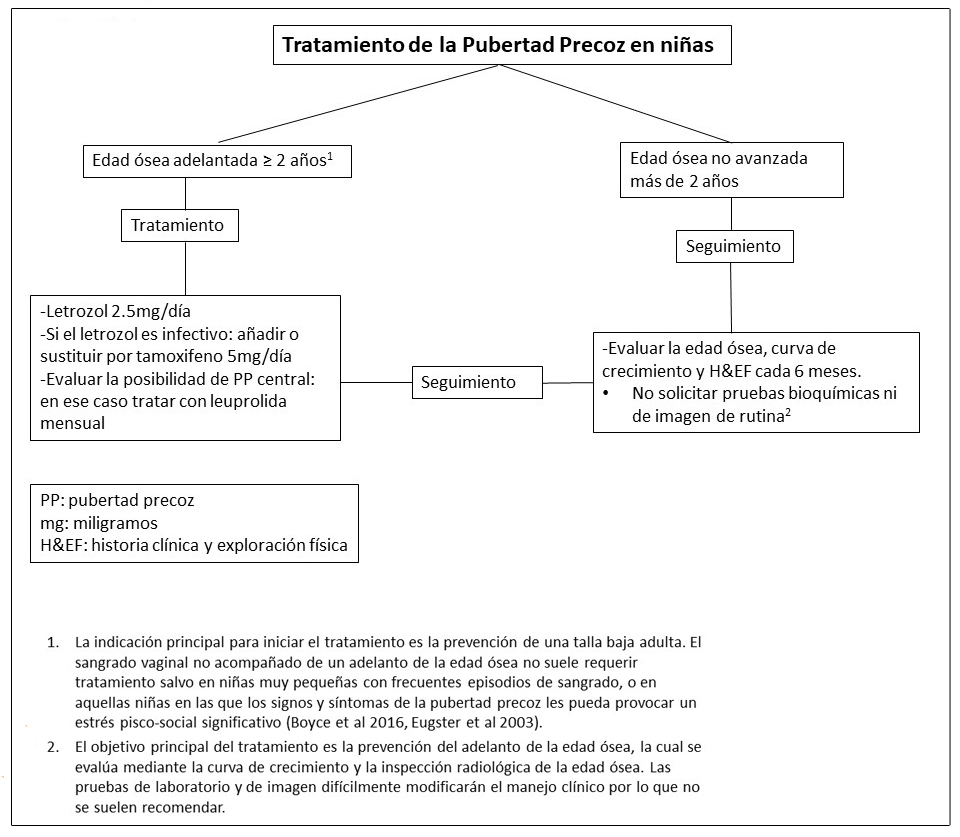
Figura 13. Manejo clínico recomendado para niños con alteraciones gonadales con Displasia Fibrosa/Síndrome de McCune-Albright
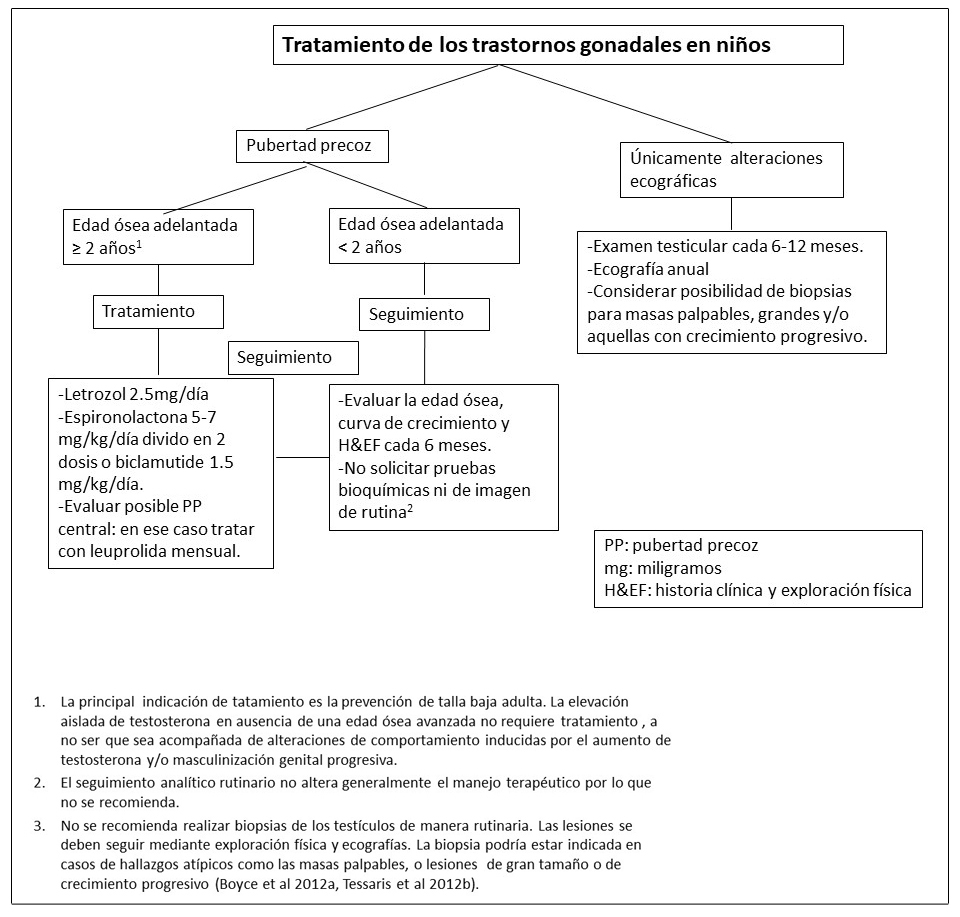
Figura 14. Recomendaciones para el manejo del hipertiroidismo en pacientes con Displasia Fibrosa/Síndrome de McCune-Albright
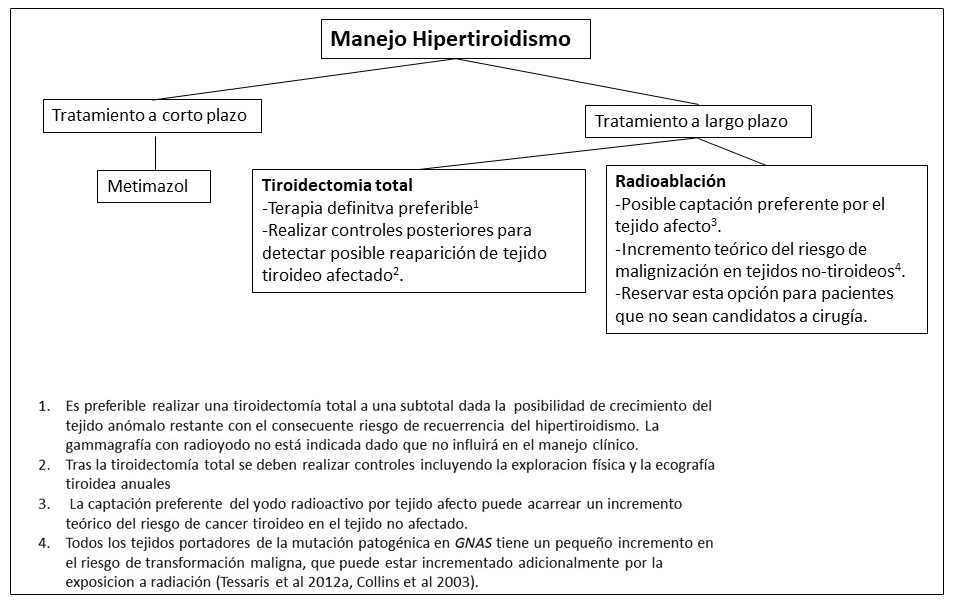
Figura 15. Recomendaciones para el manejo del exceso de hormona de crecimiento en pacientes con Displasia Fibrosa/Síndrome de McCune-Albright
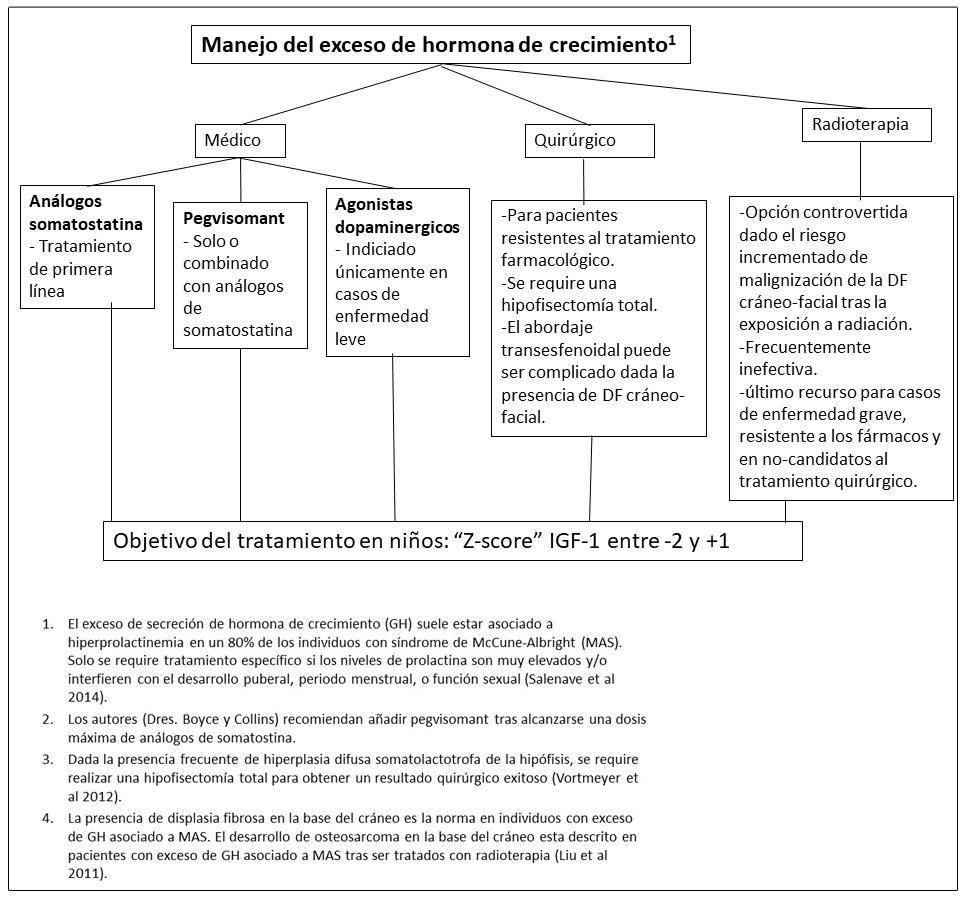
Figura 16. Recomendaciones para el manejo del exceso del hipercortisolismo en pacientes con Displasia Fibrosa/Síndrome de McCune-Albright
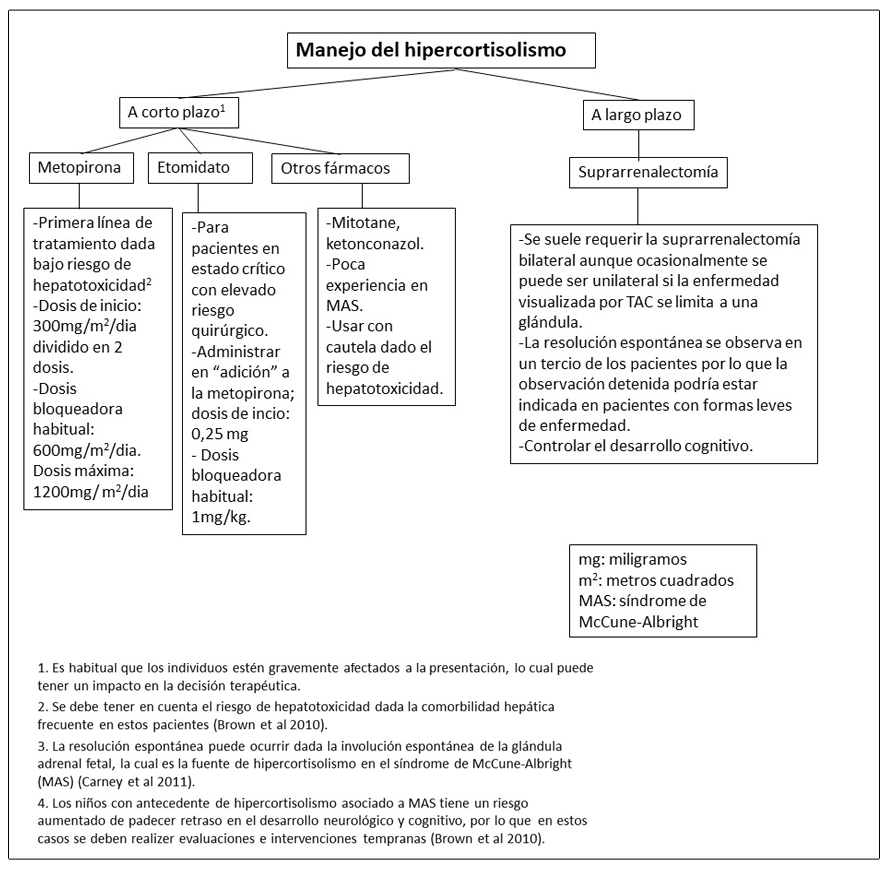
Figura 17. Recomendaciones para el manejo de alteraciones pancreáticas en pacientes con Displasia Fibrosa/Síndrome de McCune-Albright
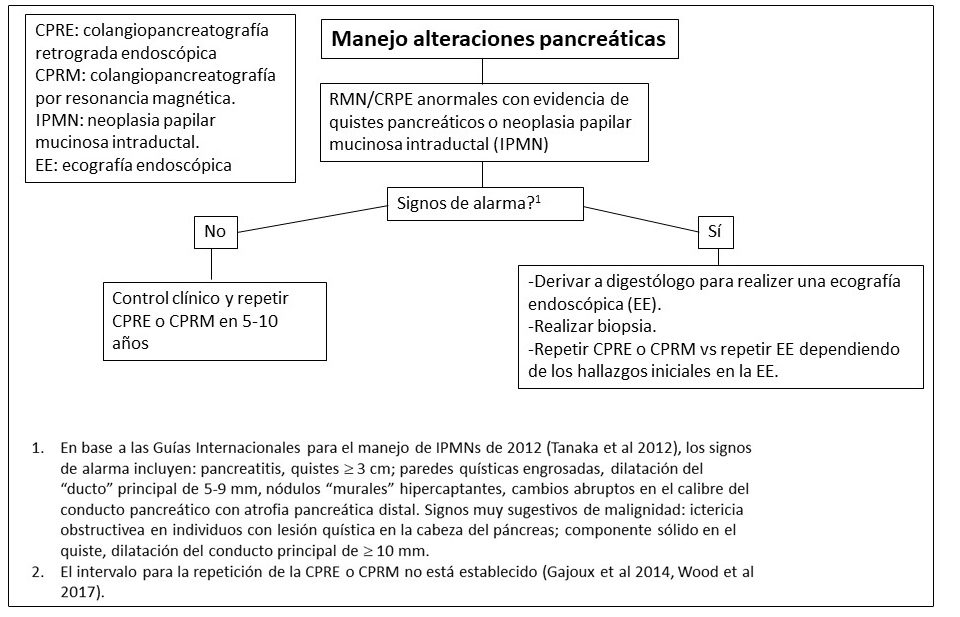
Tabla 5. Tratamiento de Otras Manifestaciones en Individuos con Displasia Fibrosa/Síndrome de McCune-Albright
- Mahdi et al. (2017), Robinson et al (2018)
- Majoor et al (2018a)
Tabla 6. Seguimiento recomendado para individuos con Displasia Fibrosa/Síndrome de McCune-Albright
Circunstancias y agentes a evitar
- Los deportes de contacto y otras actividades que entrañen un alto riesgo físico deberían evitarse en aquellos individuos con gran afectación esquelética.
- Evitar la descompresión profiláctica del nervio óptico (ver Tratamiento de las Manifestaciones).
- La extirpación quirúrgica de quistes ováricos se debe realizar en circunstancias muy específicas y con gran cautela.
- La radioterapia no esta indicada para el tratamiento de la DF. La exposición a radiación debe estar limitada en general en pacientes con DF dado el riesgo potencial para la transformación maligna (Ruggieri et al 1994).
- La ablación con yodo radioactivo también se debe evitar dado la captación preferencial por tejido tiroideo portador de la mutación lo cual podría incrementar el riesgo de malignidad en el tejido tiroideo sano.
Manejo durante el embarazo
Si bien es cierto que los efectos del embarazo sobre el hueso y las enfermedades endocrinas en mujeres con DF/MAS no han sido bien estudiadas, según la experiencia de los autores la mayor parte de las mujeres con DF que se embarazan, no sufren un empeoramiento de las manifestaciones asociadas a la enfermedad.
Terapias en Investigación
Buscar en ClinicalTrials.gov para pacientes en EEUU y en www.ClinicalTrialsRegister.eu en aquellos que vivan en Europa. Estas páginas contienen información sobre ensayos clínicos para una gran variedad de enfermedades.
NOTA: es posible que en el momento en el que usted consulte estas páginas, no haya ensayos clínicos activos para la DF/MAS.
Consejo Genético
El consejo genético es el proceso en el que se proporciona información a individuos afectos y familiares sobre la naturaleza, los patrones de herencia genética, e implicaciones de una determinada alteración genética para ayudarles a tomar decisiones personales y médicas informadas. La siguiente sección no pretende abordar todas las cuestiones personales, culturales, o éticas que un individuo pueda presentar en relación a su condición ni tampoco puede sustituir la visita con un genetista profesional.
Patrón de Herencia Genética
La Displasia fibrosa/Síndrome de McCune-Albright no se heredan:
- No hay ningún caso descrito de transmisión vertical.
- La información molecular disponible indica que todos los individuos afectos son mosaicos para la mutación GNAS la cual se origina durante el desarrollo embrionario temprano.
Riesgo a otros familiares
Padres del probando: No se ha descrito ningún caso en el que los padres de un individuo con DF/MAS presenten manifestaciones típicas de la enfermedad. Tampoco son hallazgos esperables dada la naturaleza somática de la enfermedad.
Hermanos y otros familiares del probando: dada que la DF/MAS se origina por una mutación somática, el riesgo que tiene un hermano y otros familiares de desarrollar la enfermedad es el mismo que el de la población general, es decir, no se requiere realizar una evaluación a los familiares del probando.
Riesgo de recurrencia: El riesgo de recurrencia de esta enfermedad en embarazos posteriores no está incrementado respecto al de la población general. No se requiere realizar una evaluación especial a los familiares del probando.
Hijos del probando: no existen casos descritos de transmisión vertical de la DF/MAS. Se especula que la transmisión vertical resultaría en la letalidad del embrión.
Test prenatal: Dado que la DF/MAS es secundaria a una mutación somática postzigótica y que no se hereda, no está indicado realizar diagnóstico prenatal para esta enfermedad.
Banco de DNA: se refiere al almacenaje de muestras de DNA típicamente extraídas de leucocitos de sangre periférica. Dado que es muy probable que la metodología empleada para el diagnóstico genético, y nuestros conocimientos sobre genética en general, mejoren en un futuro, podría ser de utilidad el almacenamiento de muestras de DNA de individuos afectos.
Genética molecular
Estructura del gen: GNAS es un locus complejo sometido a impronta genética (genetic imprinting) con 4 exones y promotores del cual derivan múltiples productos génico (Weinstein et al 2004). El locus cromosómico es 20q13.32. El producto génico principal de GNAS, derivado del exón 1, es la subunidad alfa de la proteína fijadora de nucleótido de guanina (Gsα) la cual está expresada en los tejidos de forma ubicua. Las entradas OMIM para el locus GNAS son 139320 y para MAS 174800.
Variantes patogénicas: Se ha identificado la mutación somática con cambio de sentido (missense mutation) p.Arg201 en más del 95% de los casos publicados de DF/MAS. Las variantes patogénicas más frecuentes son p.Arg201His y p.Arg201Cys (Lumbroso et al 2004). Más raramente, la arginina es sustituida por serina, glicina o leucina. También se ha reportado, aunque de manera excepcional, la variante patogénica p.Gln227 (Idowu et al 2007).
Hay métodos experimentales que se están desarrollando para aumentar la sensibilidad de identificación de las variantes patogénicas e incluso la cuantificación del porcentaje de células mutadas en una muestra (Bianco et al 2000, Narumi et al 2013, de Sanctis et al 2017) (ver Tabla 7). Estos métodos podrían incluso facilitar el diagnóstico en muestras de leucocitos de sangre periférica.
Tabla 7. Técnicas disponibles para la Detección de Variantes Patogénicas en pacientes con Displasia Fibrosa/Síndrome de McCune-Albright
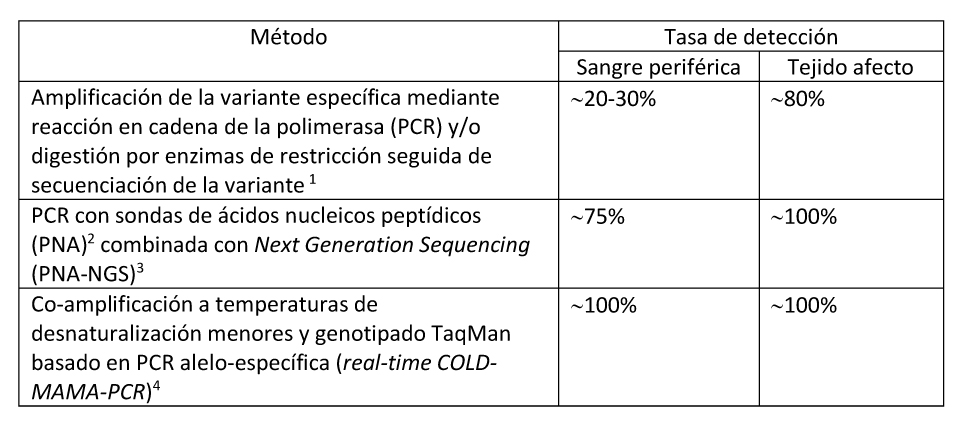
- La capacidad de detección de la variante depende del nivel de mosaicismo en el tejido y de la sensibilidad de la técnica empleada. La tasa de detección más elevada de la variante p.Arg201 usando PCR convencional se da órganos endocrinos, mientras que la más baja se da en muestras de piel. Lumbroso et al 2004, Kalfa et al 2006
- Bianco et al 2000
- Narumi et al 2013
- De Sanctis et al 2017
Estudios recientes han implicado a variantes alternativas de transcripción de GNAS en la patogénesis de DF/MAS. Se ha identificado una variante, p.Arg543His, en el producto de transcripción XLαs de Gsα (correspondería a la posición p.Arg201His en Gsα), en individuos con variantes patogénicas de origen paterno. También se ha identificado una mutación en la proteína secretora neuroendocrina (NESP55), que es otro producto de transcripción de GNAS, en tejidos afectos de individuos con variantes patogénicas de origen materno. Se han realizado estudios funcionales in vitro en XLα natural que han demostrado que esta variante de GNAS tiene la capacidad de inducir la actividad de la adenil ciclasa de manera efectiva, sugiriendo que esta variante podría contribuir a la patogénesis de la DF (Mariot el al 2011).
Tabla 8. Variantes de GNAS Somáticas Comentadas en esta Revisión
Producto genético normal: GNAS codifica la proteína Gsα la cual está asociada a la vía del AMP cíclico (cAMP). Gsα es un componente clave de muchas vías de señalización hormonal. Su rol principal es el de acoplar la proteína G con la adenil ciclasa resultando en la producción intracelular de cAMP. En su estado inactivo, la proteína Gsα forma un heterotrímero con las subunidades Gsß y Gsy, el cual va unido a una molecula de GDP. Tras la unión del ligando al receptor acoplado a la proteína G, se produce una sustitución de GDP a GTP lo cual resulta en la disociación de Gsα de las otras subunidades y en la interacción con la adenil ciclasa para inducir la producción de cAMP. Un mecanismo de actividad GTPasa hidroliza el GTP a GDP resultando en el cese de la generación de cAMP y en el reensamblaje del heterotrímero α-ß-y.
Producto genético anómalo: Las variantes patogénicas asociadas a la DF/MAS del gen GNAS p.Arg201 y p.Gln227 alteran la actividad GTPasa de Gsα causando la hiperactividad de la proteína y la producción excesiva de cAMP (Landis et al 1989).
Las manifestaciones clínicas del DF/MAS varían desde hallazgos incidentales asintomáticos a la letalidad neonatal. El fenotipo de la DF/MAS es un reflejo del rol de Gsα en ese tejido y de los tejidos portadores de la mutación. La distribución de los tejidos afectos es un reflejo del momento del desarrollo embrionario en el que ocurrió la mutación y en la capacidad de diferenciarse en diferentes tejidos de la célula mutada. Es probable que las células madre de algunos tejidos no toleren la mutación de Gsα y se eliminen durante el desarrollo embrionario. Por ejemplo, se especula que, aunque la señalización por Gsα sea muy importante en el cartílago de crecimiento, este tejido seguramente elimine las células mutadas ya que casi nunca está afectado en DF/MAS.
Las mutaciones de GNAS asociadas a la DF/MAS son el resultado de mutaciones somáticas postzigóticas por lo que es una enfermedad que existe exclusivamente el estado mosaico. Se especula que, si esta mutación existiera a nivel germinal, es decir, si todas las células estuvieran afectadas, resultaría en la letalidad embrionaria.
Cáncer y tumores benignos
Las variantes de GNAS asociadas a DF/MAS p.Arg201 y p.Gln221 (conocidas como el oncogén gsp) se han descrito en tumores benignos (Landis et al 1989) y malignos (Wood et al 2007). No obstante, la presencia aislada de estas variantes de GNAS es insuficiente para la transformación maligna de tejidos afectos, aunque probablemente predisponga para la aparición de nuevas mutaciones a nivel genético y/o epigenético.
Referencias
- Amit M, Collins MT, FitzGibbon EJ, Butman JA, Fliss DM, Gil Z. Surgery versus watchful waiting in patients with craniofacial fibrous dysplasia–a meta-analysis. PLoS One. 2011;6:e25179. [PMC free article]
- Benhamou J, Gensburger D, Chapurlat R. Transient improvement of severe pain from fibrous dysplasia of bone with denosumab treatment. Joint Bone Spine. 2014;81:549–50.
- Bhattacharyya N, Wiench M, Dumitrescu C, Connolly BM, Bugge TH, Patel HV, Gafni RI, Cherman N, Cho M, Hager GL, Collins MT. Mechanism of FGF23 processing in fibrous dysplasia. J Bone Miner Res. 2012;27:1132–41.
- Bianco P, Riminucci M, Majolagbe A, Kuznetsov SA, Collins MT, Mankani MH, Corsi A, Bone HG, Wientroub S, Spiegel AM, Fisher LW, Robey PG. Mutations of the GNAS1 gene, stromal cell dysfunction, and osteomalacic changes in non-McCune-Albright fibrous dysplasia of bone. J Bone Miner Res. 2000;15:120–8.
- Boyce AM, Brewer C, DeKlotz TR, Zalewski CK, King KA, Collins MT, Kim HJ. Association of hearing loss and otologic outcomes with fibrous dysplasia. JAMA Otolaryngol Head Neck Surg. 2018;144:102–7. [PMC free article]
- Boyce AM, Chong WH, Shawker TH, Pinto PA, Linehan WM, Bhattacharryya N, Merino MJ, Singer FR, Collins MT. Characterization and management of testicular pathology in McCune-Albright syndrome. J Clin Endocrinol Metab. 2012a;97:E1782–90. [PMC free article]
- Boyce AM, Chong WH, Yao J, Gafni RI, Kelly MH, Chamberlain CE, Bassim C, Cherman N, Ellsworth M, Kasa-Vubu JZ, Farley FA, Molinolo AA, Bhattacharyya N, Collins MT. Denosumab treatment for fibrous dysplasia. J Bone Miner Res. 2012b;27:1462–70. [PMC free article]
- Boyce AM, Glover M, Kelly MH, Brillante BA, Butman JA, Fitzgibbon EJ, Brewer CC, Zalewski CK, Cutler Peck CM, Kim HJ, Collins MT. Optic neuropathy in McCune-Albright syndrome: effects of early diagnosis and treatment of growth hormone excess. J Clin Endocrinol Metab. 2013;98:E126–34. [PMC free article]
- Boyce AM, Kelly MH, Brillante BA, Kushner H, Wientroub S, Riminucci M, Bianco P, Robey PG, Collins MT. A randomized, double blind, placebo-controlled trial of alendronate treatment for fibrous dysplasia of bone. J Clin Endocrinol Metab. 2014;99:4133–40. [PMC free article]
- Brown RJ, Kelly MH, Collins MT. Cushing syndrome in the McCune-Albright syndrome. J Clin Endocrinol Metab. 2010;95:1508–15. [PMC free article]
- Carney JA, Young WF, Stratakis CA. Primary bimorphic adrenocortical disease: cause of hypercortisolism in McCune-Albright syndrome. Am J Surg Pathol. 2011;35:1311–26. [PMC free article]
- Celi FS, Coppotelli G, Chidakel A, Kelly M, Brillante BA, Shawker T, Cherman N, Feuillan PP, Collins MT. The role of type 1 and type 2 5′-deiodinase in the pathophysiology of the 3,5,3′-triiodothyronine toxicosis of McCune-Albright syndrome. J Clin Endocrinol Metab. 2008;93:2383–9. [PMC free article]
- Clark TJ, Tan BK, Kennedy CR. Asynchronous ovarian torsion in a patient with McCune-Albright syndrome. J Obstet Gynaecol. 2000;20:204.
- Collins MT, Chebli C, Jones J, Kushner H, Consugar M, Rinaldo P, Wientroub S, Bianco P, Robey PG. Renal phosphate wasting in fibrous dysplasia of bone is part of a generalized renal tubular dysfunction similar to that seen in tumor-induced osteomalacia. J Bone Miner Res. 2001;16:806–13.
- Collins MT, Kushner H, Reynolds JC, Chebli C, Kelly MH, Gupta A, Brillante B, Leet AI, Riminucci M, Robey PG, Bianco P, Wientroub S, Chen CC. An instrument to measure skeletal burden and predict functional outcome in fibrous dysplasia of bone. J Bone Miner Res. 2005;20:219–26.
- Collins MT, Sarlis NJ, Merino MJ, Monroe J, Crawford SE, Krakoff JA, Guthrie LC, Bonat S, Robey PG, Shenker A. Thyroid carcinoma in the McCune-Albright syndrome: contributory role of activating Gs alpha mutations. J Clin Endocrinol Metab. 2003;88:4413–7.
- Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. 2012. Orphanet J Rare Dis. [PMC free article]
- Cox JL, Cushman-Vokoun AM, McGarry SV, Kozel JA. Two cases of Mazabraud syndrome and identification of a GNAS R201H mutation by next-generation sequencing. Virchows Arch. 2017;470:589–93.
- Cutler CM, Lee JS, Butman JA, FitzGibbon EJ, Kelly MH, Brillante BA, Feuillan P, Robey PG, DuFresne CR, Collins MT. Long-term outcome of optic nerve encasement and optic nerve decompression in patients with fibrous dysplasia: risk factors for blindness and safety of observation. Neurosurgery. 2006;59:1011–7.
- de Sanctis L, Galliano I, Montanari P, Matarazzo P, Tessaris D, Bergallo M. Combining real-time COLD- and MAMA-PCR TaqMan techniques to detect and quantify R201 GNAS mutations in the McCune-Albright syndrome. Horm Res Paediatr. 2017;87:342–9.
- Estrada A, Boyce AM, Brillante BA, Guthrie LC, Gafni RI, Collins MT. Long-term outcomes of letrozole treatment for precocious puberty in girls with McCune-Albright syndrome. Eur J Endocrinol. 2016;175:477–83. [PMC free article]
- Eugster EA, Rubin SD, Reiter EO, Plourde P, Jou HC, Pescovitz OH. Tamoxifen treatment for precocious puberty in McCune-Albright syndrome: a multicenter trial. J Pediatr. 2003;143:60–6.
- Feuillan P, Calis K, Hill S, Shawker T, Robey PG, Collins MT. Letrozole treatment of precocious puberty in girls with the McCune-Albright syndrome: a pilot study. J Clin Endocrinol Metab. 2007;92:2100–6.
- Florenzano P, Pan KS, Brown SM, Paul SM, Kushner H, Guthrie LC, de Castro LF, Collins MT, Boyce AM. Age-Related Changes and Effects of Bisphosphonates on Bone Turnover and Disease Progression in Fibrous Dysplasia of Bone. J Bone Miner Res. 2019 Apr;34(4):653-660.
- Ganda K, Seibel MJ. Rapid biochemical response to denosumab in fibrous dysplasia of bone: report of two cases. Osteoporos Int. 2014;25:777–82.
- Gaujoux S, Salenave S, Ronot M, Rangheard AS, Cros J, Belghiti J, Sauvanet A, Ruszniewski P, Chanson P. Hepatobiliary and pancreatic neoplasms in patients with McCune-Albright syndrome. J Clin Endocrinol Metab. 2014;99:E97–101.
- Hansen MR, Moffat JC. Osteosarcoma of the skull base after radiation therapy in a patient with McCune-Albright syndrome: case report. Skull Base. 2003;13:79–83. [PMC free article]
- Hart ES, Kelly MH, Brillante B, Chen CC, Ziran N, Lee JS, Feuillan P, Leet AI, Kushner H, Robey PG, Collins MT. Onset, progression, and plateau of skeletal lesions in fibrous dysplasia and the relationship to functional outcome. J Bone Miner Res. 2007;22:1468–74.
- Idowu BD, Al-Adnani M, O’Donnell P, Yu L, Odell E, Diss T, Gale RE, Flanagan AM. A sensitive mutation-specific screening technique for GNAS1 mutations in cases of fibrous dysplasia: the first report of a codon 227 mutation in bone. Histopathology. 2007;50:691–704.
- Ikawa Y, Yachi Y, Inoue N, Kato A, Okajima M, Yachie A. Neonatal McCune-Albright syndrome with giant cell hepatitis. J Pediatr. 2016;178:298.
- Kalfa N, Philibert P, Audran F, Ecochard A, Hannon T, Lumbroso S, Sultan C. Searching for somatic mutations in McCune-Albright syndrome: a comparative study of the peptidic nucleic acid versus the nested PCR method based on 148 DNA samples. Eur J Endocrinol. 2006;155:839–43.
- Katznelson L, Laws ER Jr, Melmed S, Molitch ME, Murad MH, Utz A, Wass JA. Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99:3933–51.
- Kelly MH, Brillante B, Collins MT. Pain in fibrous dysplasia of bone: age-related changes and the anatomical distribution of skeletal lesions. Osteoporos Int. 2008;19:57–63.
- Kelly MH, Brillante B, Kushner H, Gehron Robey P, Collins MT. Physical function is impaired but quality of life preserved in patients with fibrous dysplasia of bone. Bone. 2005;37:388–94.
- Kuznetsov SA, Cherman N, Riminucci M, Collins MT, Robey PG, Bianco P. Age-dependent demise of GNAS-mutated skeletal stem cells and “normalization” of fibrous dysplasia of bone. J Bone Miner Res. 2008;23:1731–40. [PMC free article]
- Lala R, Andreo M, Pucci A, Matarazzo P. Persistent hyperestrogenism after precocious puberty in young females with McCune-Albright syndrome. Pediatr Endocrinol Rev. 2007;4 Suppl 4:423–8.
- Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340:692–6.
- Lawless ST, Reeves G, Bowen JR. The development of thyroid storm in a child with McCune-Albright syndrome after orthopedic surgery. Am J Dis Child. 1992;146:1099–102.
- Lee JS, FitzGibbon EJ, Chen YR, Kim HJ, Lustig LR, Akintoye SO, Collins MT, Kaban LB. Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet J Rare Dis. 2012;7 Suppl 1:S2. [PMC free article]
- Lee JS, FitzGibbon E, Butman JA, Dufresne CR, Kushner H, Wientroub S, Robey PG, Collins MT. Normal vision despite narrowing of the optic canal in fibrous dysplasia. N Engl J Med. 2002;347:1670–6.
- Leet AI, Boyce AM, Ibrahim KA, Wientroub S, Kushner H, Collins MT. Bone-grafting in polyostotic fibrous dysplasia. J Bone Joint Surg Am. 2016;98:211–9. [PMC free article]
- Leet AI, Chebli C, Kushner H, Chen CC, Kelly MH, Brillante BA, Robey PG, Bianco P, Wientroub S, Collins MT. Fracture incidence in polyostotic fibrous dysplasia and the McCune-Albright syndrome. J Bone Miner Res. 2004a;19:571–7.
- Leet AI, Magur E, Lee JS, Wientroub S, Robey PG, Collins MT. Fibrous dysplasia in the spine: prevalence of lesions and association with scoliosis. J Bone Joint Surg Am. 2004b;86-A(3):531–7.
- Lim YH, Ovejero D, Sugarman JS, Deklotz CM, Maruri A, Eichenfield LF, Kelley PK, Jüppner H, Gottschalk M, Tifft CJ, Gafni RI, Boyce AM, Cowen EW, Bhattacharyya N, Guthrie LC, Gahl WA, Golas G, Loring EC, Overton JD, Mane SM, Lifton RP, Levy ML, Collins MT, Choate KA. Multilineage somatic activating mutations in HRAS and NRAS cause mosaic cutaneous and skeletal lesions, elevated FGF23 and hypophosphatemia. Hum Mol Genet. 2014;23:397–407. [PMC free article]
- Liu F, Li W, Yao Y, Li G, Yang Y, Dou W, Zhong D, Wang L, Zhu X, Hu H, Zhang J, Wang R, Chen G. A case of McCune-Albright syndrome associated with pituitary GH adenoma: therapeutic process and autopsy. J Pediatr Endocrinol Metab. 2011;24:283–7.
- Lumbroso S, Paris F, Sultan C., European Collaborative Study. Activating Gsalpha mutations: analysis of 113 patients with signs of McCune-Albright syndrome–a European Collaborative Study. J Clin Endocrinol Metab. 2004;89:2107–13.
- Mahdi AJ, Connor P, Thakur I. McCune-Albright syndrome-associated bone marrow failure and extramedullary haematopoeisis secondary to fibrous dysplasia. Br J Haematol. 2017;178:179.
- Majoor BC, Boyce AM, Bovée JV, Smit VT, Collins MT, Cleton-Jansen AM, Dekkers OM, Hamdy NA, Dijkstra PS, Appelman-Dijkstra NM. Increased risk of breast cancer at a young age in women with fibrous dysplasia. J Bone Miner Res. 2018a;33:84–90.
- Majoor BCJ, Andela CD, Quispel CR. Rotman M2, Dijkstra PDS, Hamdy NAT, Kaptein AA, Appelman-Dijkstra NM. Illness perceptions are associated with quality of life in patients with fibrous dysplasia. Calcif Tissue Int. 2018b;102:23–31. [PMC free article]
- Mancini F, Corsi A, De Maio F, Riminucci M, Ippolito E. Scoliosis and spine involvement in fibrous dysplasia of bone. Eur Spine J. 2009;18:196–202. [PMC free article]
- Manjila S, Zender CA, Weaver J, Rodgers M, Cohen AR. Aneurysmal bone cyst within fibrous dysplasia of the anterior skull base: continued intracranial extension after endoscopic resections requiring craniofacial approach with free tissue transfer reconstruction. Childs Nerv Syst. 2013;29:1183–92.
- Mariot V, Wu JY, Aydin C, Mantovani G, Mahon MJ, Linglart A, Bastepe M. Potent constitutive cyclic AMP-generating activity of XLαs implicates this imprinted GNAS product in the pathogenesis of McCune-Albright syndrome and fibrous dysplasia of bone. Bone. 2011;48:312-20. [PMC free article]
- Narumi S, Matsuo K, Ishii T, Tanahashi Y, Hasegawa T. Quantitative and sensitive detection of GNAS mutations causing McCune-Albright syndrome with next generation sequencing. PLoS One. 2013;8:e60525. [PMC free article]
- Ovejero D, Lim YH, Boyce AM, Gafni RI, McCarthy E, Nguyen TA, Eichenfield LF, DeKlotz CM, Guthrie LC, Tosi LL, Thornton PS, Choate KA, Collins MT. Cutaneous skeletal hypophosphatemia syndrome: clinical spectrum, natural history, and treatment. Osteoporos Int. 2016;27:3615–26.
- Parvanescu A, Cros J, Ronot M, Hentic O, Grybek V, Couvelard A, Levy P, Chanson P, Ruszniewski P, Sauvanet A, Gaujoux S. Optic neuropathy in McCune-Albright syndrome: effects of early diagnosis and treatment of growth hormone excess. JAMA Surg. 2014;149:858–62.
- Paul SM, Gabor LR, Rudzinski S, Giovanni D, Boyce AM, Kelly MR, Collins MT. Disease severity and functional factors associated with walking performance in polyostotic fibrous dysplasia. Bone. 2014;60:41–7. [PMC free article]
- Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey P. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112:683–92. [PMC free article]
- Robinson C, Boyce AM, Estrada A, Kleiner DE, Mathew R, Stanton R, Frangoul H, Collins MT. Bone marrow failure and extramedullary hematopoiesis in McCune-Albright syndrome. Osteoporos Int. 2018;29:237–41.
- Ross DS, Burch HB, Cooper DS, Greenlee MC, Laurberg P, Maia AL, Rivkees SA, Samuels M, Sosa JA, Stan MN, Walter MA. 2016 American Thyroid Association guidelines for diagnosis and management of hyperthyroidism and other causes of thyrotoxicosis. Thyroid. 2016;26:1343–1421.
- Ruggieri P, Sim FH, Bond JR, Unni KK. Malignancies in fibrous dysplasia. Cancer. 1994;73:1411–24.
- Salenave S, Boyce AM, Collins MT, Chanson P. Acromegaly and McCune-Albright syndrome. J Clin Endocrinol Metab. 2014;99:1955–69. [PMC free article]
- Silva ES, Lumbroso S, Medina M, Gillerot Y, Sultan C, Sokal EM. Demonstration of McCune-Albright mutations in the liver of children with high gammaGT progressive cholestasis. J Hepatol. 2000;32:154–8.
- Stanton RP, Ippolito E, Springfield D, Lindaman L, Wientroub S, Leet A. The surgical management of fibrous dysplasia of bone. Orphanet J Rare Dis. 2012;7 Suppl 1:S1. [PMC free article]
- Tanaka M, Fernández-del Castillo C, Adsay V, Chari S, Falconi M, Jang JY, Kimura W, Levy P, Pitman MB, Schmidt CM, Shimizu M, Wolfgang CL, Yamaguchi K, Yamao K., et al. International consensus guidelines 2012 for the management of IPMN and MCN of the pancreas. Pancreatology. 2012;12:183–97.
- Tessaris D, Corrias A, Matarazzo P, De Sanctis L, Wasniewska M, Messina MF, Vigone MC, Lala R. Thyroid abnormalities in children and adolescents with McCune-Albright syndrome. Horm Res Paediatr. 2012a;78:151–7.
- Tessaris D, Matarazzo P, Mussa A, Tuli G, Verna F, Fiore L, Lala R. Combined treatment with bicalutamide and anastrazole in a young boy with peripheral precocious puberty due to McCune-Albright Syndrome. Endocr J. 2012b;59:111–17.
- Vortmeyer AO, Gläsker S, Mehta GU, Abu-Asab MS, Smith JH, Zhuang Z, Collins MT, Oldfield EH. Somatic GNAS mutation causes widespread and diffuse pituitary disease in acromegalic patients with McCune-Albright syndrome. J Clin Endocrinol Metab. 2012;97:2404–13. [PMC free article]
- Weinstein LS, Liu J, Sakamoto A, Xie T, Chen M. Minireview: GNAS: normal and abnormal functions. Endocrinology. 2004;145:5459–64.
- Wood LD, Noë M, Hackeng W, Brosens LA, Bhaijee F, Debeljak M, Yu J, Suenaga M, Singhi AD, Zaheer A, Boyce A, Robinson C, Eshleman JR, Goggins MG, Hruban RH, Collins MT, Lennon AM, Montgomery EA. Patientes with McCune-Albright syndrome have a broad spectrum of abnormalities in the gastrointestinal tract and pancreas. Virchows Arch. 2017;470:391–400. [PMC free article]
- Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13.
Para obtener más información sobre nuestra guía médica o solicitar acceso a recursos especializados, contacta con nosotros a través de nuestra página de contacto.